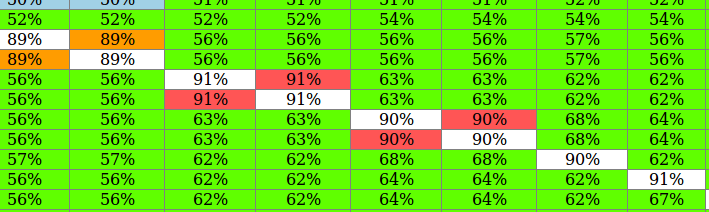
I don't know which method is used to compute the "Simple similarity" as I couldn't find any documentation except for issue UGENE-1020. Nevertheless, I believe that it's impossible for a sequence not to bear 100% identity to itself, regardless of the method used in the calculation (see picture attached). As it happens with the "include" gap option, I guess that the gaps are being computed as a difference without checking if both sequence share that gap:
a- AAT--GG
b- AAT--GG
c- CATAAGG
In that example, I guess the gaps in a and b are being computed as differences, while they shouldn't, as a and b are identical. The algorithm works for pairwise alignment, but not for a multiple alignment.
BTW, it should be called "Identity" instead of "Simple similarity" if for example a and c have a 57% identity (4/7).
- blocks
-
UGENE-1374 MSA: add distance matrix options
-

- Closed
-