-
Type:
Bug
-
Status: Closed
-
Priority:
 Major
Major
-
Resolution: Cannot Reproduce
-
Affects Version/s: 40.1
-
Fix Version/s: 51
-
Component/s: None
-
Labels:None
-
Environment:
Windows10
-
Affect Type:Userdefined
Scenario:
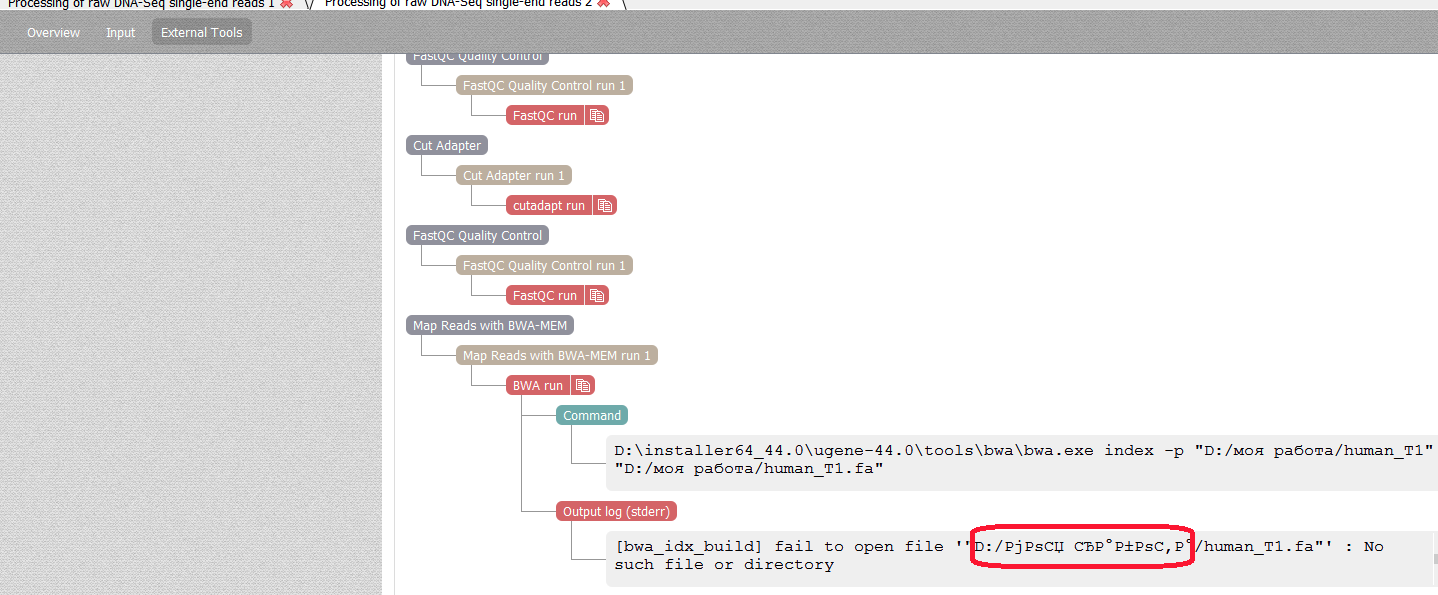
- Create directory "Моя работа"
- Put in it samples/FASTAQ/eas.fastq and samples/FASTA/human_T1.fasta
- Open Tools->NGS data analyses->"Raw DNA-Seq data processing"
- Select "Read FASTQ Files with Reads" and set parameter=eas.fastaq
- Select "Map Reads with BWA-MEM" and set Reference Genome=human_T1.fasta
- Run
Expected result: workflow finished without error
Actual result:
[ERROR][16:22] Subtask {DnaAssemblyMultiTask} is failed: Subtask {Align short reads} is failed: Subtask {Build Bwa index} is failed: Subtask {BWA tool} is failed: BWA tool exited with code 1Open external tool tab, see on the attachment that path is wrong.
Note, that workflow finished without any error, if put these 2 files in the directory without space "Моя_работа" or in the directory with space but without Russian letters "My work".